<style>
.reveal {
font-size: 30px;
}
.reveal h3{
color: #eb3474;
}
.reveal p {
text-align: left;
}
.reveal ul {
display: block;
}
.reveal ol {
display: block;
}
</style>
# <font color="#218d9e">Introduction on QDR4 with LAMMPS</font>
2021-07-30
Michael Ting-Chang Yang
楊庭彰
mike.yang@twgrid.org
Academia Sinica Grid-computing Centre (ASGC)
Last Modified in 2021-09-17
---
# Introduction of DiCOS System
* Brief Introduction:
- https://www.twgrid.org/wordpress/index.php/dicos-accelerate-research-pipeline/
* DiCOS WEB Portal:
- https://dicos.grid.sinica.edu.tw/
---
# DiCOS Registration
----
### Registration on DiCOS System
* DiCOS registration
- Go to: http://canew.twgrid.org
- Follow: Account -> Apply Account
- Apply Account:
- Academia Sinica Member: Academia Sinica Single Sign-On (AS-SSO)
- Non-AS Member: register with DiCOS account directly
* After registration, you will need to wait for the approval of your group leader to activate your group (you will be notified via e-mail)
----
### DiCOS Registration -- Step 1

----
### DiCOS Registration -- Step 2
---
# Introduction of HPC QDR4
----
### Introduction of QDR4 - Computing
* Computing Machine Specifications
- Computing Nodes: 50
- as-wn652 to as-wn682, as-wn701, as-wn709 to as-wn718, as-wn720 to as-wn727
- Local Slurm Submission Only
- Specifications of Each Node:
- CPU: Intel(R) Xeon(R) CPU E5-2650L v2 @ 1.70GHz
- CPU Cores: 20 (10 cores x2 sockets)
- Memory: 125GB
- Workflow management system (WFMS): slurm
----
### Introduction of QDR4 - Computing
* Slurm Partitions (Queues)
- Partition in QDR4 as of 2021-07-27
| PARTITION | TIMELIMIT | NODES | PRIORITY | NODELIST | TYPE |
|:-----------:|:-----------:|:-----:|:--------:|:--------:|:----:|
| large | 14-00:00:00 | 42 | 10 | as-wn[652-682,701,709-718 | CPU |
| long_serial | 14-00:00:00 | 5 | 10 | as-wn[720-724] | CPU |
| short | 3-01:00:00 | 50 | 1 | as-wn[652-682,701,709-718,720-727] | CPU |
| development | 1:00:00 | 1 | 1000 | as-wn727 | CPU/COMPILE |
| v100 | 14-00:00:00 | 1 | 1 | hp-teslav01 | GPU |
| a100 | 14-00:00:00 | 1 | 1 | hp-teslaa01 | GPU |
----
### Introduction of QDR4 - Storage
* Software repositories (readonly)
- /cvmfs/cvmfs.grid.sinica.edu.tw
* Assuming your user name is **jack**, then you have following storage spaces to use:
- Home Directory (NFS): __/dicos_ui_home/jack__
- Working Directory (CEPH): __/ceph/sharedfs/users/j/jack/__
- Group Directory (CEPH, specially for JCT group): __/ceph/sharedfs/groups/JCT__
* You will be prompted with the relative information when login into slurm-ui01
* * Utilization Dashboard:
- http://dicos-dashboard.twgrid.org/hpc_qdr4
- Group leader will have a specific dashboard of the resource usage of group members (work in progress)
----
### Introduction of QDR4 - Network
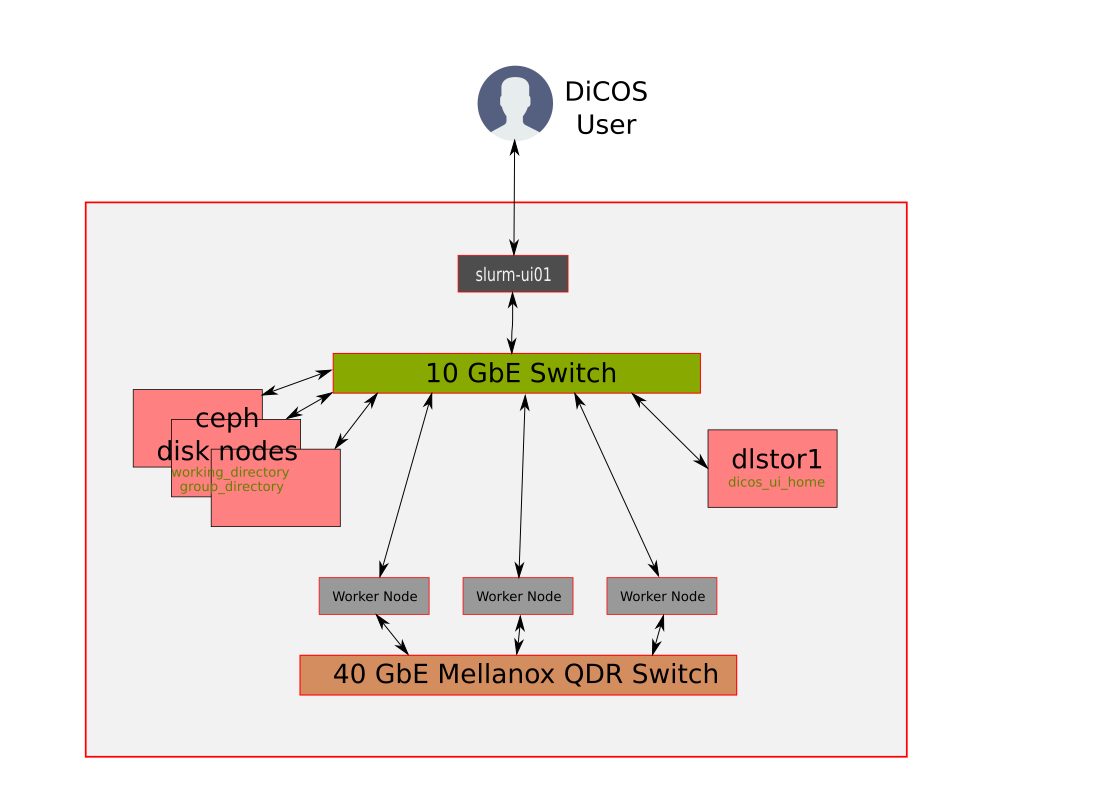
---
# User Interface (Login Node) and User Information
----
### Login into QDR4 User Interface
* The user interface node for QDR4 is **slurm-ui01.twgrid.org**
```bash
ssh jack@slurm-ui01.twgrid.org
```
* You will be prompted with the relative information of your account when login into slurm-ui01
* System Utilization Dashboard:
- We are making a utilization dashboard now, and will announce to users when it is ready
---
# Basic Usage of Slurm System
----
### Basic Usage of Slurm System
* Query cluster information
```bash
sinfo
```
* Query the jobs submitted by you
```bash
sacct
```
or
```bash
sacct -u jack
```
----
### Basic Usage of Slurm System
* Submit your job with bash script (recommended)
- You need to add __#!/bin/bash__ in the first line of your script
```bash
sbatch your_script.sh
```
* Submit your job (binary executable) with srun
- It's not easy to control the resource usage with direct submit with **srun**. We will recommand you to wrap your **srun** command within a batch shell and then run with **sbatch**
```bash
srun your_program arg1 arg2
```
* Show queue information
```bash
squeue
```
----
### Basic Usage of Slurm System
* Show your job in the queue
```bash
squeue -u jack
```
* Show the detailed job information
```bash
scontrol show job your_jobid
```
* Cancel your job
```bash
scancel your_jobid
```
---
# Environment Modules
----
### Environment Modules - Use Scopes
* QDR4: slurm system
- The environment modules will be initialized automatically when you login in UI
- You could load the necessary modules in UI and then submit your job, the environment settings will be bring to worker nodes automatically
* FDR5: condor system
- The environment modules will be initialized automatically when you login in UI
- You could load environment modules in UI for testing
- But if you would like to submit job to worker nodes and use the environment modules. You will need to source initialize script of modules with your script to enable the modules functionality
----
### Environment Modules
* Detailed information please refer to the original document:
- https://modules.readthedocs.io/en/latest/
* Environment-modules help user to setup environment and envoronment variables properly for specific software environments
- User doesn't need to worry about the complex settings of the environments
* Show available modules in QDR4
```less
$ module avail
------ /cvmfs/cvmfs.grid.sinica.edu.tw/hpc/modules/modulefiles/Core ---------
app/anaconda3/4.9.2 app/cmake/3.20.3 app/root/6.24 gcc/9.3.0 gcc/11.1.0
intel/2018 nvhpc_sdk/20.11 python/3.9.5 app/binutils/2.35.2 app/make/4.3
gcc/4.8.5 gcc/10.3.0 intel/2017 intel/2020 pgi/20.11
```
----
### Basic Usage of Environment Modules
* Load module
```bash
module load intel/2020
```
* Unload module
```bash
module unload intel/2020
```
* Show currently loaded modules
```bash
module list
```
* Unload all loaded modules
```bash
module purge
```
---
# Building and Using LAMMPS in QDR4
* Assuming your input file is **SSMD_input.txt**
* <h6>Originally in the demonstration session it is:</h6>
- <h6>SSMD_jc20210527a00_antiSym_FdChkAll_round_CS_SS_HardP_BiMix_U0.1_dt1e-5_1e8_Rdm_Rf10.0.txt</h6>
* Assuming your input file is in you home directory
* **$HOME/SSMD_input.txt**
* i.e. if you are jack: **/dicos_ui_home/jack/SSMD_input.txt**
---
# MPI-enabled LAMMPS (Intel + Make)
----
### MPI-enabled LAMMPS (Intel + Make)
* Building LAMMPS with **Intel Compiler** and **Make**
* Load Modules
```bash
module load intel/2020
module load mpich
```
* Build LAMMPS (+GRANULAR package)
```bash
mkdir lammps-src
cd lammps-src
wget https://download.lammps.org/tars/lammps-3Mar2020.tar.gz
tar zxvf lammps-3Mar2020.tar.gz
cd lammps-3Mar20/src
make yes-GRANULAR
make ps # Show your make package selections
make serial # A. Make serial version of LAMMPS
make icc_mpich # B. Make icc+mpich version of LAMMPS
```
----
### MPI-enabled LAMMPS (Intel + Make)
* Run LAMMPS locally (in slurm-ui01)
```bash
cp ~/SSMD_input.txt .
./lmp_icc_mpich -in SSMD_input.txt
```
---
# Note on the Demonstration
* However, I will suggest you to build with **cmake**, which will
- Generate Makefile according to your requirements
- Show the building progress during make (build) stage
- I will show how to build LAMMPS with **cmake** in the following slides
- LAMMPS need to build with cmake3
---
# MPI-enabled LAMMPS (Intel + CMake)
----
### MPI-enabled LAMMPS (Intel + CMake)
* Load Intel 2020 and cmake3 modules
```bash
module load intel/2020
module load mpich
module load app/cmake/3.20.3
```
----
### MPI-enabled LAMMPS (Intel + CMake)
* Generate a Makefile with **cmake** and assign the installation directory of LAMMPS to **$HOME/lammps**
```bash
mkdir lammps-src
cd lammps-src
wget https://download.lammps.org/tars/lammps-3Mar2020.tar.gz
tar zxvf lammps-3Mar2020.tar.gz
cd lammps-3Mar20
mkdir build
cd build
export INTEL_PATH=/cvmfs/cvmfs.grid.sinica.edu.tw/hpc/compiler/centos7_intel_2020
cmake -DCMAKE_C_COMPILER=$INTEL_PATH/compilers_and_libraries_2020.4.304/linux/bin/intel64/icc \
-DCMAKE_CXX_COMPILER=$INTEL_PATH/compilers_and_libraries_2020.4.304/linux/bin/intel64/icpc \
-DMPI_CXX_COMPILER=$INTEL_PATH/oneapi/mpi/2021.2.0/bin/mpiicpc \
-DMPI_CXX=$INTEL_PATH/oneapi/mpi/2021.2.0/bin/mpiicpc \
-DMPI_CXX_LIBRARIES=$INTEL_PATH/oneapi/mpi/2021.2.0/lib/libmpicxx.so \
-D BUILD_MPI=yes \
-D BUILD_OMP=yes \
-D MPI_CXX_INCLUDE_PATH=$INTEL_PATH/oneapi/mpi/2021.2.0/include/ \
-D PKG_GRANULAR=yes \
-D CMAKE_INSTALL_PREFIX=$HOME/lammps \
../cmake
```
----
### MPI-enabled LAMMPS (Intel + CMake)
* Make (Compile) and install LAMMPS in **slurm-ui01**
```bash
make
make install
```
* Make (Compile) and install LAMMPS by submitting a slurm job (**recommended**)
- **-o**: standard output to **make.log**
- **-e**: standard error to **make.err**
```bash
srun -o make.log -e make.err make &
make install
```
----
### MPI-enabled LAMMPS (Intel + CMake)
* Then the LAMMPS will be installed in **$HOME/lammps**, make LAMMPS program (lmp) globally discoverable, use:
```bash
export PATH=$HOME/lammps/bin:$PATH
```
* Create job directory (lammps-job) and copy input files
```bash
mkdir lammps-job
cd lammps-job
cp ~/SSMD_input.txt .
```
----
### MPI-enabled LAMMPS (Intel + CMake)
* Run LAMMPS in **serial mode** (in slurm-ui01 locally, **not recommended**)
```bash
lmp -in SSMD_input.txt
```
* Run LAMMPS in **MPI mode** (in slurm-ui01 locally, **not recommended**)
```bash
mpirun -np 3 lmp -in SSMD_input.txt
```
* Note: you should not use local MPI run on slurm-ui01
----
### MPI-enabled LAMMPS (Intel + CMake)
* Run LAMMPS with Example Inputs (**SLURM+MPI**, **not recommended**)
- Note: I will **not recmmend** to use this example. Because we could not control very well on the claimed resources, which will cause overclaim on the same recources if there is more job submitted to slurm.
```bash
srun -N 1 mpirun -np 3 lmp -in SSMD_input.txt
```
----
### MPI-enabled LAMMPS (Intel + CMake)
* Run LAMMPS with Example Inputs (**SLURM+MPI**, **RECOMMENDED**)
- Write a bash script with the contents (named as myscript.sh)
```bash
#!/usr/bin/bash
#SBATCH --job-name=jcttest # shows up in the output of 'squeue'
#SBATCH --time=1-00:00:00 # specify the requested wall-time
#SBATCH --nodes=3 # -n number of nodes allocated for this job
#SBATCH --ntasks-per-node=1 # number of MPI ranks per node
#SBATCH --cpus-per-task=3 # -c number of OpenMP threads per MPI rank
#SBATCH --error=job.%J.err # job error. By default, both files are directed to a file of the name slurm-%j.err
#SBATCH --output=job.%J.out # job output. By default, both files are directed to a file of the name slurm-%j.out
module purge
module load intel/2020
module load mpich
module load lammps/jct/3Mar2020
## create mpi_work in your HOME directory and copy these scripts from /ceph/astro_phys/user_document/scripts/
srun mpirun lmp -in SSMD_input.txt
```
----
### MPI-enabled LAMMPS (Intel + CMake)
- Run the script with sbatch:
```bash
sbatch myscript.sh
```
---
# Using Pre-built LAMMPS with Slurm + MPI
----
### Using Pre-built LAMMPS with Slurm + MPI
* I have made a customized pre-built LAMMPS for JCT lab
* The version built in June, 2021 is without USER-OMP package
- The older version have no OPENMP enabled function
- I have built new LAMMPS for JCT group with OPENMP function (USER-OMP package)
* Current available built LAMMPS commands in the pre-built packages
```less
lmp lmp_PL1_Vc600 lmp_PL1_Vc6e-2
lmp_PL1_Vc6 lmp_PL1_Vc6000 lmp_PL1_Vc6e-3
lmp_PL1_Vc60 lmp_PL1_Vc6e-1 lmp_PL1_Vc6e-5
```
----
### Using Pre-built LAMMPS with Slurm + MPI
* Run LAMMPS with **Slurm + MPI** (**RECOMMENDED**)
- User are recommended to use sbatch instead of srun to submit the job
- Example bash shell myscript.sh:
```bash
#!/usr/bin/bash
#SBATCH --job-name=JCT_TEST # shows up in the output of 'squeue'
#SBATCH --time=1-00:00:00 # specify the requested wall-time
#SBATCH --nodes=3 # -n number of nodes allocated for this job
#SBATCH --ntasks-per-node=1 # number of MPI ranks per node
#SBATCH --cpus-per-task=1 # -c number of OpenMP threads per MPI rank
#SBATCH --error=job.%J.err # job error. By default, both files are directed to a file of the name slurm-%j.err
#SBATCH --output=job.%J.out # job output. By default, both files are directed to a file of the name slurm-%j.out
module purge
module load intel/2020
module load mpich
module load lammps/jct/3Mar2020
export OMP_NUM_THREADS=3
srun lmp -sf omp -pk omp 3 -in SSMD_input_run.txt
```
----
### Using Pre-built LAMMPS with Slurm + MPI
- Run with sbatch:
```bash
sbatch myscript.sh
```
---
# Note on the example script
----
### Note on the example script
* LAMMPS MPI job could be run intra-node (same node) or inter-node (across nodes):
- **--nodes=3**: use 3 nodes for MPI jobs
- **--ntasks-per-node=3**: 3 MPI job per node
- **--cpus-per-task=3**: Claim OPENMP using 3 cores for ecch MPI job
* So for **--nodes=3 --ntasks-per-node=3 --cpus-per-task=3** will claim 27 CPU cores across 3 nodes
* If your are going to use USER-OMP (openmp) feature, you need to assign
- **OMP_NUM_THREADS=3** is the environment variables ask OPENMP to use 3 cores per threads for calculation
- **lmp -sf omp -pk omp 3** is tell lmp to use OPENMP feature and 3 cores per threads for calculation
- You need to use the settings above togehter with identical **--cpus-per-task** setting to get correct resource claim
```bash
export OMP_NUM_THREADS=3
srun lmp -sf omp -pk omp 3 -in SSMD_input_run.txt
```
---
# Computation Time of SSMD_input.txt
----
### Computation Time of SSMD_input.txt
* Reduce the input for Run 1 only
| serial | nodes | ntasks-per-node | cpus-per-task | mean CPU usage per task | time | cores |
|:------:|:-------:|:---------------:|:-------------:|:-----------------------:|:----------:|:----:|
| 1 | 1 | 1 | 1 | 99.4% | 0:03:19 | 1 |
| 2 | 1 | 1 | 3 | 290.0% | 0:02:42 | 3 |
| 3 | 1 | 3 | 1 | 99.4% | 0:02:29 | 3 |
| 4 | 3 | 1 | 1 | 50.6% | 0:05:05 | 3 |
| 5 | 3 | 3 | 3 | 58.8% | 0:03:53 | 27 |
----
### Computation Time of SSMD_input.txt
* The result is a bit weird for me, but maybe you could check the LAMMPS document for tuning
- From document:
- https://docs.lammps.org/Run_basics.html
- If you run LAMMPS in parallel via mpirun, you should be aware of the processors command, which controls how MPI tasks are mapped to the simulation box, as well as mpirun options that control how MPI tasks are assigned to physical cores of the node(s) of the machine you are running on. These settings can improve performance, though the defaults are often adequate.
- We need to check more detail with the document for the usage of LAMMPS
---
# Dedicated Partition (Queue) for Compilations
* A dedicated partition (queue) is available for compilation and short running jobs
- Partition:
- Name: development
- Time limit: 1 day per job
---
# LAMMPS with Condor (FDR5)
* The workload management system of FDR5 is the same to IoP PC1
* We have made a slide for FDR5 condor job submissions, you could refer to https://docs.twgrid.org/p/Obn3p7srm#/
---
# Problem Report and FAQ
----
### Problem Report and FAQ
* Online documents: https://dicos.grid.sinica.edu.tw/wiki/slurm_docs/
* On site document: /ceph/astro_phys/user_document/
* On site shell template: /ceph/astro_phys/user_document/scripts/
* Email channel to ASGC admins: Dicos-Support@twgrid.org
* Regular face-to-face (live) concall: ***ASGC DiCOS User Meeting*** (held at 13:20 (UTC+8), every Wednesday), ask our staffs for the concall information
{"title":"Introduction on QDR4 with LAMMPS","tags":"presentation,DiCOS_Document,JCT","type":"slide","slideOptions":{"transition":"fade","theme":"black","parallaxBackgroundImage":"/uploads/upload_76121c8a0e0fa435cd31d35029943729.jpg"}}